Seit zwei bis drei Jahren verzeichnet die Branche der Cannabis Hersteller einen neuen Hype – den Ausblick auf die medizinische Anwendung, insbesondere von THC-haltigen Produkten. Die positive Wirkung von Cannabis und der daraus abgeleiteten Produkte in der Medizin ist unbestritten; ob als Schmerzmittel, bei Epilepsie oder anderen krankheitsbedingten Krampfzuständen. Aber auch mit Blick auf die Suchtbehandlung ist die Legalisierung von Cannabis weltweit auf dem Vormarsch. Damit eröffnen sich für die Hersteller ganz neue Möglichkeiten, vor allem im Segment der qualitativ hochwertigen Arzneimittel, die dem aktuellen Preisverfall im Segment der CBD-Produkte entgegenwirken könnten.
Doch der positiven Aussicht stehen gleichzeitig neue Herausforderungen entgegen. Die Herstellung eines Arzneimittels und des zugehörigen Arzneiwirkstoffs unterliegt strengen gesetzlichen Regelungen; insbesondere Qualitätsanforderungen an Produkt, Prozess und Verarbeitung, die gemeinhin unter dem Begriff der „Guten Herstellungspraxis“ (engl. GMP – Good Manufacturing Practice) zusammengefasst werden. Dabei handelt es sich hier nicht um ein „einfaches und freiwillig“ umzusetzendes Qualitätssicherungssystem.
Vielmehr geht es um eine Fülle von detaillierten Anforderungen, die sich an Themen wie Personal, Hygiene, Rohstoffe, technische Einrichtungen, Dokumentation, Prozesse, Analysen und vieles mehr richten. Festgeschrieben sind diese Anforderungen – bei Herstellung in der Schweiz – sowohl in den Europäischen als auch in den PIC/S Guidelines. Weitere zu beachtende GMP-Richtlinien kommen hinzu, will man seine Produkte in Länder exportieren, die außerhalb des Geltungsbereiches der EU und PIC/S liegen.
Wird die Produktion auf medizinisches Cannabis ausgerichtet, muss man sich bereits sehr früh mit dieser Thematik beschäftigen. Einer der häufigsten Fehler besteht darin, dass schnell mit den ersten Einrichtungen, dem Auf- und Ausbau von Produktionsstätten, begonnen wird, ohne die dafür im GMP-regulierten Umfeld geforderten speziellen Planungsunterlagen erstellt zu haben. Dies aber kann am Ende das gesamte Projekt gefährden, wenn es um die behördliche Inspektion und Zertifizierung geht.
Was dich auch interessieren könnte...


Am Anfang stehen die Produktidee und natürlich der Businessplan – beides zunächst scheinbar ohne Verbindung zu GMP. Aber weit gefehlt! Gerade die Überlegungen zum Produktportfolio, den Bezugsquellen (z. B. Biomasse oder Blüten) und den geplanten Märkten bestimmen ganz wesentlich die regulatorischen und GMP-Anforderungen. Es geht um Fragen des Imports, Exports und der GMP-Compliance möglicher ausländischer Lieferanten. All das muss genauestens überlegt und in der Strategie festgelegt werden.
Ganz am Anfang muss geklärt werden, was – in diesem regulatorisch noch sehr unsicheren Umfeld – wirklich machbar ist und was nicht. Schon manche sind darüber gestolpert, dass ihr Businesspartner in Kanada nicht EU GMP-fähig war oder der mazedonische Hersteller aus regulatorischer Sicht keine Erlaubnis für die Belieferung des deutschen Marktes hatte. Ein GMP-Compliance Check der möglichen Partner ist hier schon recht früh gefragt.
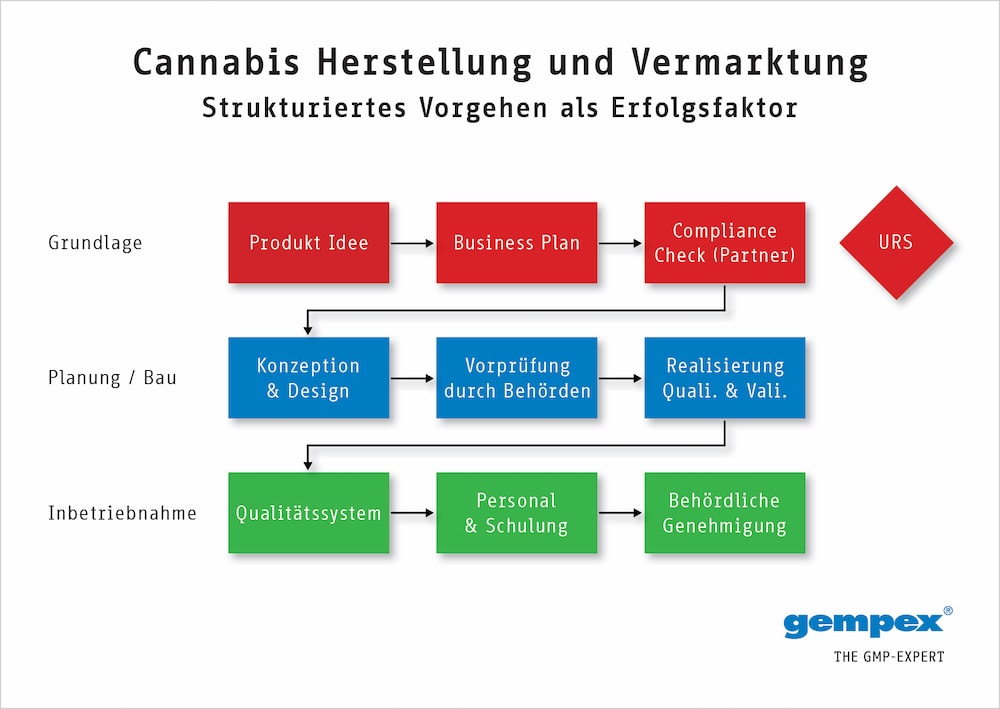
Abbildung 1 zeigt ein sehr vereinfachtes Schema der wichtigsten Schritte in dem sequenziell vorgeschriebenen Ablauf. Nur wer diese Schritte konsequent einhält, hat auch eine Chance auf die am Ende notwendige Zertifizierung.
Ein im GMP-Umfeld wichtiges und auch behördlich gefordertes Dokument sind die User Requirement Specifications (URS) – kurz: die Nutzeranforderungen. In diesem Dokument werden die Ergebnisse der Businessstrategie (allerdings ohne die wirtschaftlichen Aspekte) festgeschrieben. Es werden darin insbesondere die aus der Strategie folgenden und zu beachtenden Basis GMP-Anforderungen fixiert. Dieses Dokument ist die Grundlage für alle weiteren Aktionen und wird während des gesamten Lebenszyklus des Projektes aktuell gehalten. Es ist ein formales Dokument, das mit Datum und Unterschrift offiziell freigegeben werden muss.
Sind die regulatorischen Randbedingungen gesetzt, kann mit der baulichen Grundkonzeption – in der Regel mit ersten Layout-Plänen – begonnen werden. Hier gilt es aber, erstmalig aus GMP-Sicht auf diese Pläne zu schauen, sogenannte Design Reviews durchzuführen. Es geht um die Einrichtung nach GMP vorgeschriebener Hygienezonen, um das Festlegen klar definierter Personal- und Materialflusspläne, dem Festlegen von Funktionsräumen entsprechend dem logischen Produktionsablauf, Luftführungs- und Luftqualitätsstrategien und um vieles mehr. Es ist also neben dem grundtypischen „Bauplan“ auch ein „GMP-Zonenplan“ gefragt, der eine wichtige Grundlage für die erste Planprüfung durch die Behörde darstellt. Das Planprüfungsgespräch ist ein nicht zu unterschätzender Projektmeilenstein. Erst nach diesem kann eigentlich der weitere Baufortschritt empfohlen werden.
In der Phase der Realisierung – Bau oder Umbau der Produktionsstätte – wird der Cannabis Hersteller mit einer neuen GMP-typischen Anforderung konfrontiert, der Qualifizierung. Diese entspricht einer sehr tiefgehenden, detaillierten und systematischen technischen Prüfung, die jedoch vorgeplant und ausführlich dokumentiert werden muss. Alle Dokumente müssen dabei in einer bestimmten Reihenfolge entstehen und von dem späteren Hersteller und einer ihm zugeordneten Qualitätsperson unterschrieben und freigegeben werden. Auch hier kann das Nicht-Einhalten einer vorgegebenen Reihenfolge das gesamte Projekt gefährden.
Es gilt, für jedes einzelne technische System eine URS zu erstellen und das System einer Risikobetrachtung zu unterziehen. Ferner müssen Prüfdokumente für die Installations-, Funktions- und Leistungsprüfung (engl. IQ, OQ, PQ – Installation, Operational-, Performance Qualification) entstehen, die Prüfungen durchgeführt und mit einem Bewertungsbericht abgeschlossen werden. Erst wenn all diese Tests durchgeführt sind und ein mängelfreier Bericht vorliegt, können die Aktivitäten zur Herstellung aufgenommen werden – mit einer Einschränkung: der Validierung. Denn ähnlich wie bei der technischen Anlage unterliegt auch der Herstellungsprozess selbst intensiven Prüfungen, die im gleichen Umfang formal beschrieben und abgearbeitet werden müssen.
Steht die Anlage, so folgt als nächster wichtiger Schritt die Einrichtung eines GMP-gerechten Qualitätssicherungssystems. GMP strebt ein hohes Maß an Produktsicherheit an und fordert daher, dass alle Vorgänge und Arbeitsschritte in einer Anweisung dokumentiert sein müssen. Das soll helfen, dass unterschiedliche Personen stets gleiche Arbeitsabläufe durchführen, dass Fehler vermieden und auch bewährte Abläufe fixiert und nicht vergessen werden. Doch nicht nur die Anweisungen zu den Abläufen sind zu erstellen. Erwartet wird, dass jeder Ablauf akkurat aufgezeichnet wird, dass Fehler erfasst, bewertet und Änderungen nur nach vorheriger Genehmigung durchgeführt werden. Es sind neben den Anweisungen also auch jede Menge Protokolle zur Aufzeichnung der Abläufe zu generieren. Manch einer übersetzt GMP auch schon mal mit einer „Ganzen Menge Papier“. 60 und mehr Anweisungen und viele weitere Dokumente und Protokolle sind hierbei keine Ausnahme.
Der letzte und sicher der schwierigste Schritt besteht in der Umsetzung all dessen, was man in den Dokumenten beschrieben hat. Und hierbei steht der Mensch, das Personal, im Mittelpunkt. Es gilt auch hier die alte Weisheit, dass ein System nur so gut ist, wie es verstanden, akzeptiert und am Ende auch umgesetzt wird. Und das ist in der Praxis eine große Herausforderung. So mancher GMP-Anwärter ist am Ende daran gescheitert, dass das eingestellte Personal viele Punkte nicht verstanden und daher auch nicht oder nicht korrekt umgesetzt, d. h. mangelhaft dokumentiert hat. Bei einem solchen Projekt kann nicht früh genug damit begonnen werden, das geplante Personal zeitig einzustellen, in den Projektablauf unmittelbar einzubinden und ständig begleitend zu schulen. Es ist sicher nicht vermessen zu sagen, dass, basierend auf eigener Erfahrung, nochmals mit ein bis zwei Jahren nach Errichtung einer Anlage gerechnet werden muss, bis alles weitgehend klaglos und fehlerfrei läuft.
Die GMP-Compliance – die Übereinstimmung mit den GMP-Anforderungen – steht dann zu guter Letzt auf dem Prüfstein, wenn es um die förmliche, behördliche Abnahme geht, die Inspektion. Hier werden nicht nur die Anlage, die festgelegten Zonen und die technischen Ausführungen geprüft. Auch das Qualitätssicherungssystem, die Qualifizierung, die Validierung und das Verhalten des Personals (die durch das Personal geführten Dokumente) werden sehr genau betrachtet. Jetzt zeigt sich, ob man das Thema GMP ernst genommen hat, ob rechtzeitig begonnen und die vorgegebene Reihenfolge eingehalten wurde. Hat man all dies zu sehr auf die leichte Schulter genommen, wird spätestens an diesem Punkt klar, was die Bedeutung von GMP ist.

Zum guten Schluss: Die Bedeutung von GMP
Viele Vorkommnisse in der Vergangenheit, bei denen Menschen durch qualitativ fehlerhafte Arzneimittel geschädigt oder gar getötet wurden, haben zu der Entwicklung der GMP-Regelwerke geführt. Es geht am Ende um den Schutz des Menschen; hier konkret eines kranken Menschen, der auf die Medizin vertraut. Dies erlaubt keinerlei Spielraum mit Blick auf eine qualitativ einwandfreie Herstellung. Aus diesem Grund fokussiert GMP mindestens auf die folgenden drei wesentlichen Aspekte:
Vermeidung jeglicher Art der Kontamination des Produktes (physikalisch, chemisch und mikrobiell) oder anderer qualitativer Beeinträchtigungen (z. B. falsche oder fehlende Wirkung). Möglichst reproduzierbare Prozesse, die zeigen, dass man den Prozess beherrscht und unter Kontrolle hat. Detaillierte und lückenlose Dokumentation, die die gleichbleibenden Prozesse unterstützt und im Falle eines Fehlers jederzeit eine Rückverfolgung und Fehlerfindung ermöglicht.

Über den Autor Ralf Gengenbach
Ralf Gengenbach, Chemieingenieur (TU Karlsruhe), gründete 2002 die gempex GmbH, ein internationales GMP-Dienstleistungsunternehmen. Davor war er als interner GMP-Berater der BASF in zahlreichen Fachausschüssen aktiv (DIN, VCI, DECHEMA) und früh in die Ausarbeitungen und Kommentierungen des PIC7S-Dokuments PI006 (Validierung) und des ICH Q7-GMP-Leitfadens involviert. Sein gesammeltes Wissen hat Ralf Gengenbach – neben zahlreichen Veröffentlichungen, Vorträgen und Vorlesungen – in dem Buch „GMP, Qualifizierung und Validierung Wirkstoffanlagen“ niedergeschrieben (erschienen im Wiley Verlag).